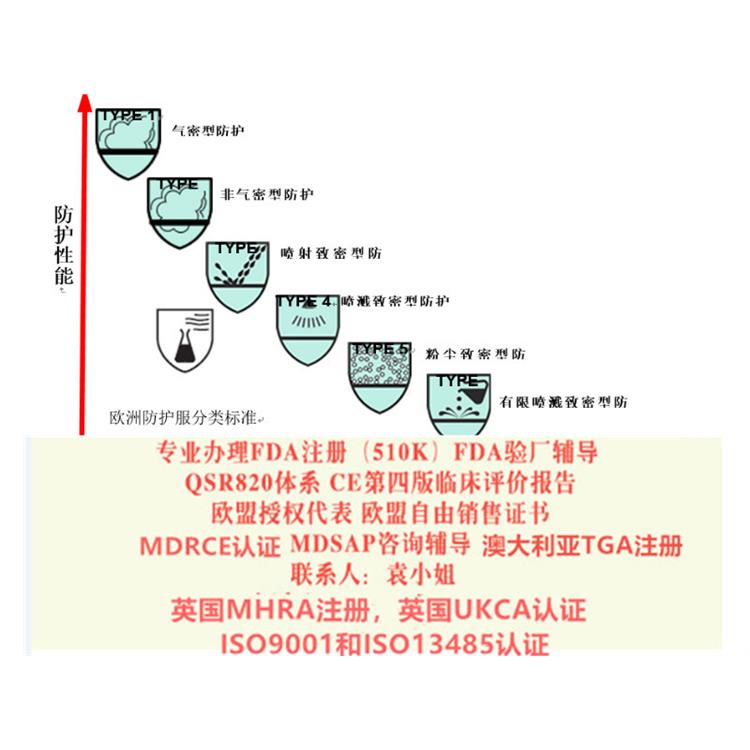
- 产品描述
英国授权代表UK Responsible Person是什么?
器械英国MHRA注册简介
英国脱欧后,按照脱欧协议,将陆续不再认可欧盟CE认证,对于器械,CE认证在英国可继续使用至2023年6月30日,但需要持有CE认证的企业在英国当地有英国负责人(类似欧盟授权代表),由英国负责人进行MHRA注册,才能进入英国GB地区市场(英格兰,威尔士和苏格兰)。2023年7月1日起,不再认可CE认证,必须进行UKCA认证。
MHRA=Medicines and Healthcare products Regulatory Agency 英国药品和产品当局
我们可以为您提供的自主服务项目主要有:
出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证
出口美国法规:器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系、食品FDA验厂及整改、OTC药品FDA验厂及整改
中国法规:器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证、SFDA验厂、SFDA注册检测、企业标准编制、局自由销售证。
出口其余国际法规:器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂、ISO22716 GMPC验厂、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试
关于经济运营商各方义务
法规在第I章第2条定义中提出了“经济运营商”的概念,经济运营商是指制造商、授权代表、进口商、经销商以及任何对系统或手术包类器械进行组合或并投放市场的自然人或法人。即在符合法规规定情况下负责器械生产(包括组合或)、销售及上市后运营的自然人或法人。
法规先规定了制造商的义务,涵盖生产、合规、上市后的产品全生命周期,但法规同时规定,经销商、进口商或其他自然人或法人在向市场提供以其名字、注册商标命名的器械时应承担制造商相应的义务,也包括变更相应器械预期用途或变更其他影响其符合性的事项的情况。在上市后要求中,经济运营商同时负有相应的责任和义务。
法规对各方义务的描述更为明确也更为具体,对于制造商的要求更为细化,因此新法规执行后,各方应先明确自身职责和义务,规范有序地开展生产和市场活动,应审核确认上游供应商是否符合规定,并确认能够自己的下游流程符合规定,应按照对应的警戒系统的要求进行或配合事件上报,配合完成现场纠正措施,并依据职责组织培训。
与高风险MD有关的新要求
• 对于III类植入器械和用于注入和/或移除的IIb类有源器械,其评价报告,除了NB审核以外,还需要由NB交至主管当局进行expertpanel review,综合意见进行审评。
• 对于III类和植入器械(顾客定制除外) , 制造商应提交Summary of safety and clinical performance至NB。经NB审核后上传至EUDA MED数据库,供公众查询。
The manufacturer shall mention
on the label or instruc- tions for
use where the summary is available .(Article 32)
说到这里 企业应该如何面对新法规的升级呢?按照MDR法规要求。关键的内容包括如下几个方面:
企业的质量管理体系 EN ISO13485:2016 ,产品的型式试验 TYPE TESTING , 产品的技术文件 TECHNICAL CONSTRUCTION FILES要满足这些要求,通常需要咨询机构和咨询师的协助。本公司拥有一支经验丰富的咨询师队伍可以为贵司提供这些服务,包括: 协助贵司建立/升级器械质量管理体系,将MDR法规的内容整合进去 协助贵司确定产品的欧盟协调标准,确认检测实验室的,样品准备以及检测不合格整改的研讨 按照MDR要求协助贵司准备技术文件,包括风险分析报告,评估资料,基本要求检查表等 协助贵司按照欧盟的要求修订说明书和标签,使其满足出口的要求。
在此基础上,会议对于MDR及IVDR的执行达成了如下时间表。2016年12月14日,EUDAMED(European databank for medical devices) 筹划会上欧盟各国对于器械法规MDR及IVDR的执行进行了一轮的讨论,与会人员对于这两个法规的细节内容进行了讨论并达成了一致意见。在此基础上,会议对于MDR及IVDR的执行达成了如下时间表。一,DMR的主要变化1.扩大了应用范围2.提出了新的概念和器械的定义3.细化了器械的分类4.完善了器械的通用和性能要求5.加强对技术文件的要求6.加强器械上市后的7.完善评价相关要求8.提出Eudamed数据库的建立和使用9.提出器械的可追溯性(UDI)10.对NB提出严格的要求MDR简介2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。MDR将取代Directives 90/385/EEC (有源植入类器械指令)and 93/42/EEC(器械指令)。
十多年里,SUNGO已为全球30多家上市公司和全球器械制造商,合计5000多家企业提供过相关服务。
SUNGO公司介绍 SUNGO GROUP: SUNGO TECHNICAL SERVICE INC美国公司; SUNGO Certification Company Limited英国公司 SUNGO Europe B.V.荷兰公司; SUNGO Cert GmbH德国公司; SUNGO Australia澳大利亚公司; 上海沙格企业管理咨询有限公司() 上海沙格企业管理咨询有限公司武汉分公司 上海沙格企业管理咨询有限公司广东分公司 SUNGO集团凭借全球网络和专业队伍为全球客户提供法规,在医疗器械行业尤为专长。 我们可以为您提供的自主服务项目主要有: 出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表、欧盟自由销售证书、防护服PPE指令Type5/6认证 出口美国法规:医疗器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂辅导及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系辅导、食品FDA验厂辅导及整改、OTC品FDA验厂辅导及整改 法规:医疗器械产品备案登记表、医疗器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证辅导、SFDA验厂辅导、SFDA注册检测、企业标准编制、监局自由销售证。 出口其余国际法规:医疗器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂辅导、ISO22716 GMPC验厂辅导、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试 上海沙格企业管理咨询公司已经在国内近千家客户提供了相关服务,也受美国几家大型采购集团委托,对其在的供应商进行二方审核工作,我们的客户集中在无纺布行业企业、敷料耗材、病床、轮椅、体外诊断医疗器械行业、大型饲料机械设备企业、电子电器行业类别。公司一直秉承“专业辅导、增值服务、、本土价格”的方针,为客户提供优质的服务。 我们服务过的部分知名企业及上市公司有: 新华医疗、鱼跃医疗、威高集团、阳普医疗、艾迪尔医疗、驼人医疗、恒安集团、康德莱集团、阿蓓纳、上海联影、上海复旦医疗、微创骨科医疗、普罗医学、凯利泰、联医医疗、羚锐制、江西3L、正昌集团、杭州可靠、台钜集团、维达集团、青岛光电、科勒、暴龙眼镜等等
欢迎来到上海沙格企业管理咨询有限公司网站,我公司位于历史文化悠久,近代城市文化底蕴深厚,历史古迹众多,有“东方巴黎”美称的上海市。 具体地址是上海浦东陆家嘴公司街道地址,负责人是袁玲。
主要经营ISO13485认证。
本公司以科技为前导,质量为中心,全员为基础,依国际水准,创智博品牌。质量、信誉是公司生存和发展的基石;ISO9000质量体系为保证。选择我们的产品:咨询 管理咨询 ,是正确的决定!
本页链接:http://www.cg160.cn/vgy-94975988.html
以上信息由企业自行发布,该企业负责信息内容的完整性、真实性、准确性和合法性。阿德采购网对此不承担任何责任。 马上查看收录情况: 百度 360搜索 搜狗
- 产品推荐
- 四川机械制造新材料检测机构 多年从业经验 江门免疫化学测试标准 华微检测 律师咨询电话 北京咨询律师免费 中山水质检测tds 详细介绍 北京劳动仲裁多长时间内申请 劳动案件律师咨询 西安油品清洁度检测标准 抑制试验 皮肤消毒剂检测流程 经验丰富 建筑工程承包合同纠纷律师咨询 法律咨询中心 宿迁自然光老化试验第三方检测机构-优尔鸿信检测 山东检测机构cross section 银川接口安全风险评估 软件检测机构 腾创 菏泽噪音测试第三方检测温湿度循环测试
- 相关文章
- 医用帽2017/745 2017/745 / EU的CE认证升级指南纸尿裤2017/745 MDR医疗器械分类规则瑞代 吸唾管瑞代要求 MDRCE辅导要求腕式血压计的英代认证要求 什么是英国授权代表医用绷带英国代表 什么情况下需要提供英代床边扶手fda认证510k 美国FDA510K认证是什么 如何申请杨克连接管UKREP认证怎么申请 英国授权代表是什么棉签美国FDA510K认证 美国FDA510K认证是什么 如何申请电动救护车担架2017/745 医疗器械MDR法规纱布块MDRCE证书怎么申请 mdrce认证 评估包含什么注射器的UKREP认证怎么申请 英国授权代表是什么口罩MDSAP认证 怎么做
关于上海沙格企业管理咨询有限公司
商铺首页 |
更多产品 |
联系方式
SUNGO公司介绍 SUNGO GROUP: SUNGO TECHNICAL SERVICE INC美国公司; SUNGO Certification Company Limited英国公司 SUNGO Europe B.V.荷兰公司; SUNGO Cert GmbH德国公司; SUNGO Australia澳大利亚公司; 上海沙格企业管理咨询有限公司() 上海沙格企业管理咨询有限公司武汉分公司 上海沙格企业管理咨询有限公司广东分..
- 我要给“电动病床的MDRCE认证生产厂家”留言
- 更多产品


















