
- 产品描述
申请国外主管当局的证书需要满足什么条件?
原则上应确保产品满足出具国/地区的法规要求,例如在欧盟高风险产品需要有CE证书,低风险产品必须行注册。
MDR及IVDR正式执行 2017年5月底
MDR强制执行 2020年5月底
IVDR强制执行 2022年5月底
欧盟授权代表(European Authorised Representative 或European Authorized Representative)是指由位于欧洲经济区的制造商明确的一个**人或法人。该**人或法人可代表EEA的制造商履行欧盟相关的指令和法律对该制造商所要求的特定的职责。
新方法指令要求欧盟授权代表必需位于欧洲经济区境内并且具有贸易注册地址;
-- EEA成员国的主管机关可以随时找上欧盟授权代表核查的制造商是否履行了欧盟相关的指令和法律所要求的职责;
制造商的一般商务代表(例如授权经销商),不论是否位于欧洲经济区境内,都不应该与的欧盟授权代表搅浑;
-- 固然欧盟授权代表可代表的制造商履行欧盟相关的指令和法律对该制造商所要求的特定的职责,但制造商依然是承担主要责任的一方。
医疗器械产品做欧盟CE认证,是要提供欧盟代表简称欧代的,如果是出口欧盟一类的你可以不用做CE认证,只要欧盟授权代表注册就行,一类灭菌测量,二类及以上才需要进行CE认证。
选择有资质,有能力的第三方欧盟授权代表公司。
欧盟境内注册的合法公司/拥有的技术人员,熟悉欧盟相关法规,能帮助制造方解决争端/避免空壳公司,代理商以及展会服务商。
------以上几点可通过查询其欧盟注册证书,拨打其欧盟境内的电话看是否为电话录音等途径进行确认。
签订有效的欧盟授权代表协议或合同。
合同/协议的甲乙双方的名称和地址必须和将来加贴CE标志的产品的包装/标签上的制造商和欧盟授权代表的名称和地址完全一致。
欧盟授权代表定义
欧盟授权代表(European Authorized Representative)是指由位于欧洲经济区EEA(包括EU与EFTA)的制造商明确的一个自然人或法人。该自然人或法人可代表EEA的制造商履行欧盟相关的指令和法律对该制造商所要求的特定的职责。
我公司办理产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德等CE认证(MDD/MDR法规),CE技术文件编订, CE*四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。
制造商的一般商务代表(例如授权经销商),不论是否位于欧洲经济区境内,都不应该与的欧盟授权代表搅浑;
-- 固然欧盟授权代表可代表的制造商履行欧盟相关的指令和法律对该制造商所要求的特定的职责,但制造商依然是承担主要责任的一方
医疗器械产品做欧盟CE认证,是要提供欧盟代表简称欧代的,如果是出口欧盟一类的你可以不用做CE认证,只要欧盟授权代表注册就行,一类灭菌测量,二类及以上才需要进行CE认证。
应注意以下事项:
欧盟授权代表合同条款应是欧盟的主要语言版本,一般为欧代所在国家的语言版本。
避免在欧盟境内无固定办公地点和固定联络电话的欧盟授权代表。
根据欧盟的法律要求,为了实现产品的可追溯行(traceability),制造商投放到欧盟市场的加贴了CE标志的产品必须标有制造商的名称和联络地址, 如果制造商来自欧洲经济区EEA(包括EU和以外的国家,其产品必须同时在标签(铭牌或包装)上标有制造商和制造商的欧盟授权代表的名称和联络地址。
为确保欧盟主管机关能适时的查验到加贴CE标志的产品的技术文件(TCF),制造商应将新版本技术文件保存在欧盟授权代表处,保存时间为自后一批产品出口后10年。
欧盟制造商的产品在欧盟出现任何故障/事故/召回等问题,应由欧盟授权代表进行联络,通报,并与主管机关进行沟通联系。
欧盟销售(Certificate of Free Sale)指的是欧盟的主管当局出具的企业产品可以在特定区域销售的文件,简称为CFS。
目前欧洲所有的CFS 只给位于其境内的公司颁发,这些公司可以是制造商、欧盟代表、 贴牌厂商。因此的企业要申请CFS证书,只能是通过其欧盟授权代表来完成。
企业申请欧盟销售证书CFS 的条件:
(1)了欧盟授权代表,签署了书面协议;
(2)产品有合法性的,这包括:
a. 如果是I 类的器械,需完成了MHRA 注册;
b.如果是I\IIA\IIB\III 类器械,获得了公告机构CE证书。
用品才可以欧盟销售证书的,欧盟有CE证书,ISO 13485证书就可以了, 中东,南美 尤其是:沙特、阿根廷、埃及 这些会要这种欧盟销售证书的,客户销售企业的产品的时候,当地要求必须注册成功才可以销售产品,那么注册的时候是需要这些文件的,MHRA颁发的销售证书,能企业生产的产品满足欧盟的法规要求,可以在欧盟市场销售。但通常欧盟不会要求企业出具CFS,只需CE证书,即可完成清关。
欧盟成员国以外的一些,比如埃及、巴西、阿根廷、印度尼西亚、委内瑞拉等会要求企业出示CFS证书。
我公司:
1. 美国代理人、FDA器械注册、FDA510K、FDA QSR820体系,
2. FDA QSR820验厂及整改、FDA警告信应对&RED LIST REMOVAL
3. TUV/NQA机构的:器械CE认证(93/42/EEC)、体外诊断器械CE认证(98/79/EC)
4.ISO9001:二015体系认证与版本升级、ISO13485:二016体系认证与版本升级
5.欧盟授权代表(EC-REP:representative in the EU)、英国MHRA器械注册
6. CFS 欧盟销售证,医进出口商会销售证书
7. GMP认证、ISO22716 GMPC验厂, 英国BRC认证,BSCI验厂
8. 验证方案设计和报告编制,器械欧盟标准检测,企业标准编制, MSDS编务
9. 口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)
10. 手术衣EN13795测试、防护服type5/6 认证和,PPE认证(89/686/EC个人防护指令)
自由销售证书(出口销售书)用途
一、在收货方海关清关中使用:执行贸易保护国家的海关要求必须出具出口销售证书、自由销售证书才能清关提货。
二、在进口国注册登记使用:进口方在本国分销销售货物产品时,出于对产品本身的安全、质量等考虑,要求出具该产品的自由销售证书并在当地质量、门注册登记后才可以在进口国自由销售该批货物。
三、对产品质量是否合格、产品是否合法生产销售的:比如向贸易方以及贸易国:该产品为质量安全、产品达到相关标准执行什么指令、产品为合法生产销售等其它
四、其它用途,如顾客提出或进口商提出。

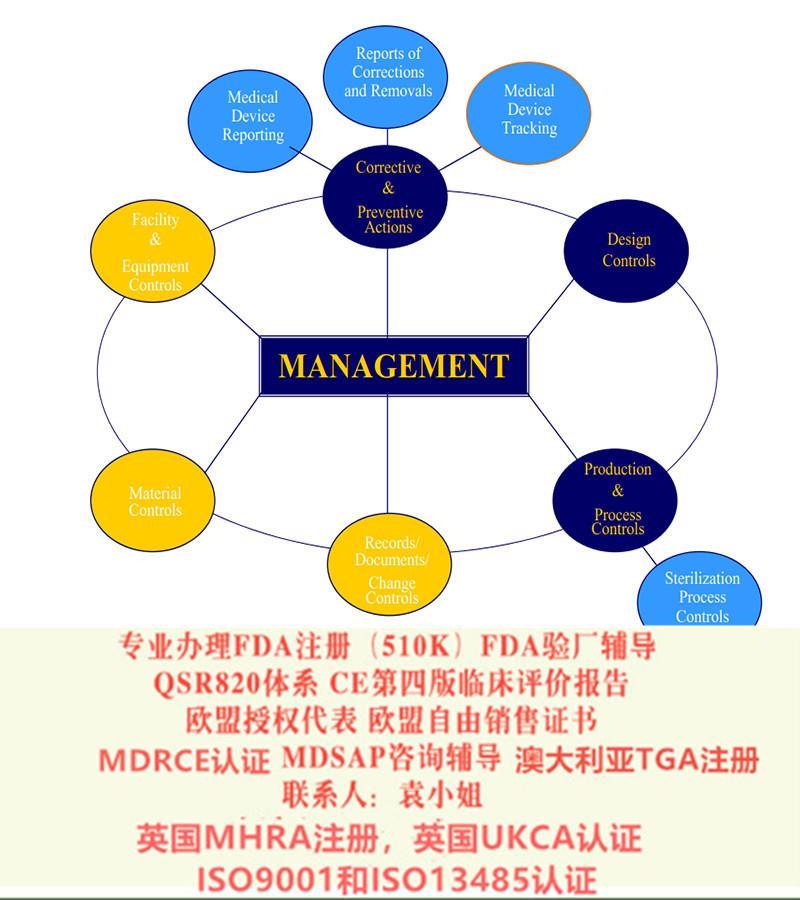

SUNGO品牌创建于2006年,立志于成为化的法规技术服务商。我司可以办理:1:欧盟MDR CE咨询,MDR欧盟授权代表,欧盟注册2:欧盟IVDR CE咨询,IVDR欧盟授权代表,欧盟注册3:美国FDA注册,FDA510K4:国内的注册证和生产证5:加拿大的MDEL注册6:ISO13485咨询和认证目前SUNGO在中国、欧洲、北美和澳洲均设有服务机构,服务过的客户较是覆盖了(中国、越南、马来西亚、孟加拉、新加坡)、欧洲(英国、瑞士、瑞典、丹麦、挪威)、北美(美国、加拿大)、南美(阿根廷)、大洋洲(澳大利亚)和非洲(博茨瓦纳、南非)等国家和地区。SUNGO致力于为的生产商和经营者提供市场准入的合规咨询以及**注册服务。从产品生产、检测、过程管理、注册、认证、整改、上市跟踪等各环节为企业提供的技术支持,为产品合规和顺利上市保驾**。十多年里,SUNGO已为30多家上市公司和制造商,合计5000多家企业提供过相关服务。SUNGO始终追求支持、服务和客户满意。所有客户都有一对一的客服对接以保持经常性的联系,提供在线即时服务,针对贸易中存在的技术壁垒方面的问题提供的支持和解。选择SUNGO,不是选择了一次性的合作伙伴,而是选择了一个长期的技术支持的伙伴。我们将秉承一贯“服务、客户”的原则,依托的技术团队,优化我们的服务,让更多的器械合法、安全进入市场,为器械行业健康发展贡献力量。因为,所以放心!
欢迎来到上海沙格企业管理咨询有限公司网站,我公司位于历史文化悠久,近代城市文化底蕴深厚,历史古迹众多,有“东方巴黎”美称的上海市。 具体地址是上海金山公司街道地址,负责人是袁小姐。
主要经营上海沙格企业管理有限公司SUNGO是欧盟授权代表,从事FDA验厂、欧盟自由销售证书、医疗器械单一体系审核MDSAP、CE*四版临床评价报告、MDRCE认证、CE MDR认证、CE技术文件、EU 201。
我们公司主要提供商务服务 认证服务 等服务,我们确信,凭借我们的专业服务和良好的协调、沟通能力,定能使客户在经营生产中无后顾之忧,协助客户不断成长,在合作中与客户实现共赢。欢迎您致电咨询!
本页链接:http://www.cg160.cn/vgy-116883603.html
以上信息由企业自行发布,该企业负责信息内容的完整性、真实性、准确性和合法性。阿德采购网对此不承担任何责任。 马上查看收录情况: 百度 360搜索 搜狗
- 产品推荐
- 化妆品注册申报 大同特殊化妆品注册手续 申报流程 上海港出口监管仓库管理 上海汇逸国际货运供应 酒吧设计工程 多样色彩巧运用 吉扩工商 大渡口公司注册团队 西安食堂装修工程 施工效率高 化妆品注册备案 揭阳特殊化妆品注册过程 申报时间 江津财务代帐服务 快速启动 吉扩工商 南开区财务外包 服务有保障 西餐厅装修施工 提升空间利用率 特殊化妆品注册 茂名进口化妆品注册 申报时间 西安设计健身会所方案 实用性强 多样色彩巧运用 特殊化妆品注册 渭南特殊化妆品注册手续 申报周期
- 相关文章
- 胰岛素注射针清洗验证方案 UKCA证书 需要什么资料隔离衣的清洗验证方案 EC Rep 需要什么资料创可贴MHRA认证 MHRA注册 的关键纸尿裤MHRA认证 医疗器械产品MHRA注册 新规要求介绍颊面管MHRA注册 英国mhra注册 mhra如何申请电动救护车担架MHRA注册 MHRA认证注册 新规要求介绍手术衣的清洗验证方案 MDR CE证书 需要什么资料眼镜自由贸易证书 欧盟自由销售证书CFS 申请材料介绍病床自由销售证书 自由贸易证书 申请条件丁腈手套自由销售证书 CFS证书 有效期多久护具CFS证书 什么是欧盟自由销售证书 申请条件尿液分析仪美国FDA注册 什么是FDA注册流程
关于上海沙格企业管理咨询有限公司
商铺首页 |
更多产品 |
联系方式
SUNGO品牌创建于2006年,立志于成为化的法规技术服务商。我司可以办理:1:欧盟MDR CE咨询,MDR欧盟授权代表,欧盟注册2:欧盟IVDR CE咨询,IVDR欧盟授权代表,欧盟注册3:美国FDA注册,FDA510K4:国内的注册证和生产证5:加拿大的MDEL注册6:ISO13485咨询和认证目前SUNGO在中国、欧洲、北美和澳洲均设有服务机构,服务过..
- 我要给“代步车CFS证书 欧盟自由销售证书CFS证书 客户清关需要”留言
- 更多产品



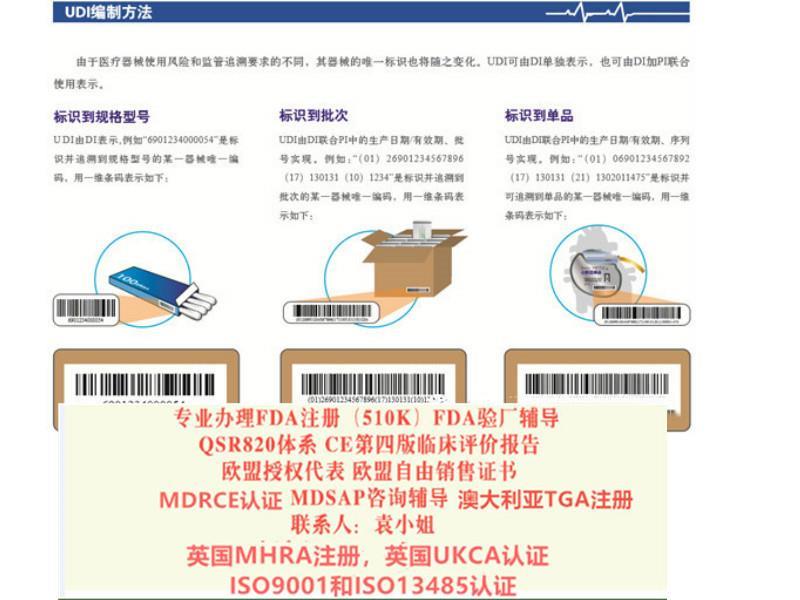








相关分类






